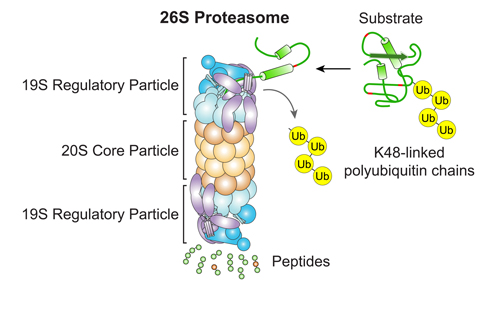
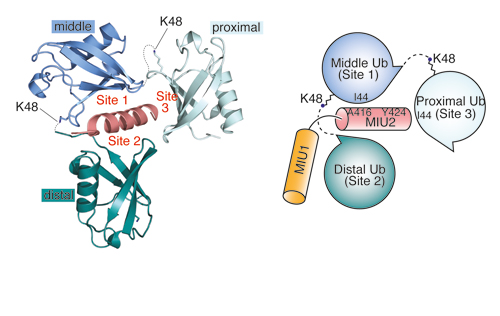
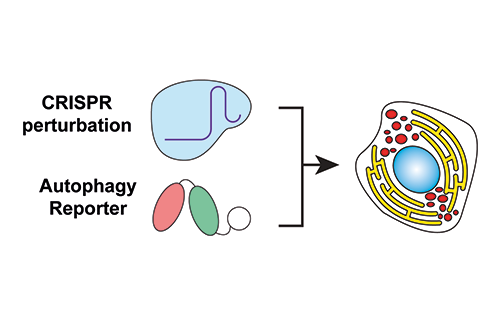
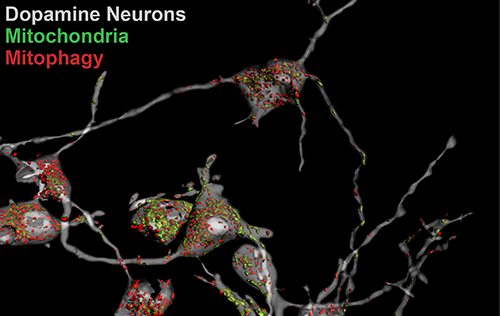
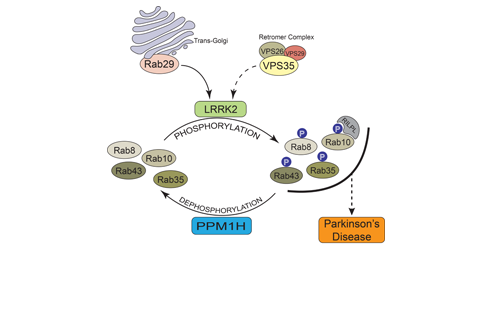
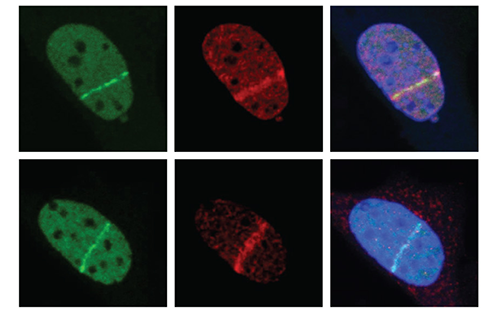
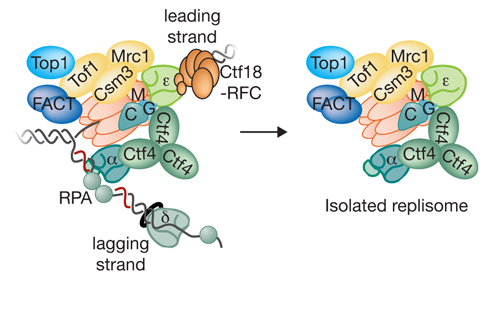
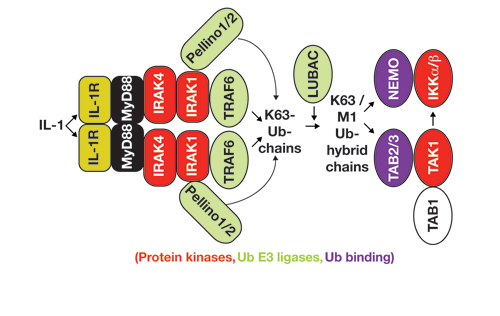
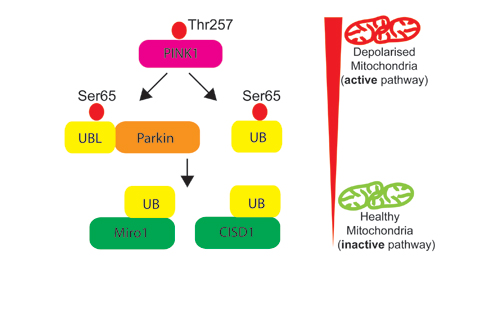
Our Research
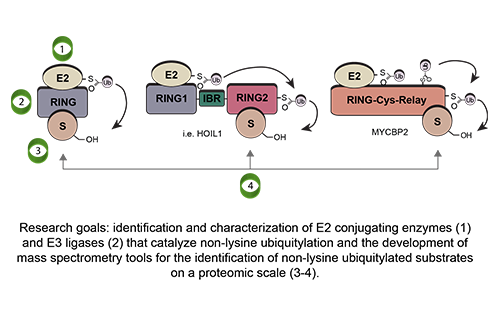
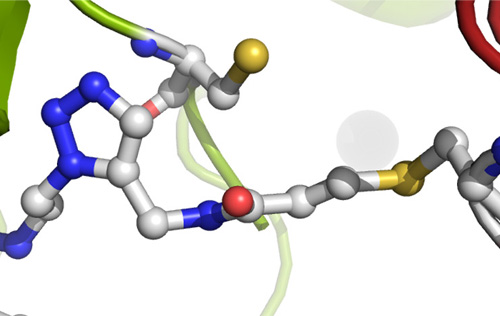
Our research groups study diverse aspects of protein ubiquitylation and phosphorylation, which regulate almost all areas of eukaryotic cell biology. Abnormalities in these pathways cause numerous diseases, and our work is focussed in particular on links to cancer, neurodegeneration and inflammation.
This is an exhilarating time to study ubiquitylation and phosphorylation, as there is still so much to discover, and the MRC PPU is uniquely placed to address these issues. We work in an unusually collaborative environment, alongside many other groups with related interests in Dundee’s Faculty of Life Sciences. We believe that the total of our efforts can be very much more than the sum of the parts.
Learning how disease can result from disruptions in phosphorylation and ubiquitin networks will reveal novel drug targets, together with improved strategies to treat these maladies in the future. This is the ultimate goal of our research, though we are also driven by sheer curiosity and a constant desire to understand more deeply how cells regulate their biology via a myriad of post-translational protein modifications.