Ayaz explains why it is hard to suppress Akt activity with PDK1 inhibitors.
Mutations leading to inappropriate activation of Akt isoforms contribute to proliferation and survival of a significant proportion of human cancers. Akt is activated by phosphorylation of its T-loop residue (Thr308) by the 3-phosphoinositide dependent kinase-1 (PDK1) and its C-terminal hydrophobic-motif (Ser473) by mTOR complex 2 (mTORC2).
A PhD student in Dario Alessi's lab, Ayaz Najafov, characterised the first selective PDK1 inhibitors such as GSK2334470 recently developed by pharmaceutical companies as potential anti-cancer agents.
However, Ayaz found frustratingly that all structurally diverse PDK1 inhibitors he tested were surprisingly ineffective at suppressing Akt activation under conditions that these compounds effectively suppressed activation of other PDK1 substrates such as S6K, RSK and SGK isoforms.
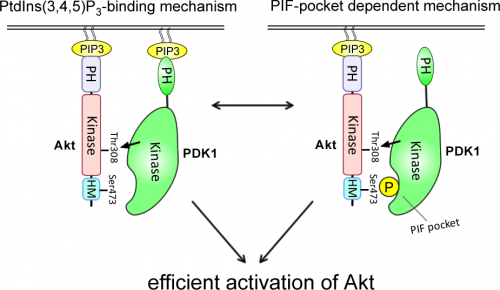
After considerable research Ayaz discovered that resistance to PDK1 inhibitors results from Akt being efficiently recruited to PDK1 via two alternative mechanisms. The first involves ability of Akt and PDK1 to mutually interact with the PI 3-kinase second messenger PtdIns(3,4,5)P3. The second entails recruitment of PDK1 to Akt after its phosphorylation at Ser473 by mTORC2, via a substrate-docking motif termed the PIF-pocket. Ayaz showed that disruption of either the PtdIns(3,4,5)P3 or the Ser473 phosphorylation/PIF-pocket mechanism, only moderately impacts on Akt activation, but induces marked sensitization to PDK1 inhibitors.
The implications of Ayaz's findings is that it will be difficult to ablate Thr308 phosphorylation and hence activation of Akt, employing PDK1 or even mTOR inhibitors as single agents. Ayaz's solution was to combine PDK1 and mTOR inhibitors. Indeed in all cell lines he tested, Ayaz observed that combinations of mTOR and PDK1 inhibitors potently suppresses Thr308 and Ser473 phosphorylation to below-basal levels also inhibiting phosphorylation of Akt substrates. Furthermore, combination of PDK1 and mTOR inhibitors had much greater effects on inhibition of proliferation of all evaluated cancer cell lines, than treatments with either of the inhibitors alone.
Ayaz's results suggests that combination of PDK1 and mTOR inhibitors might represent a new therapeutic strategy for treatment of cancers that harbour mutations elevating Akt activity. To read Ayaz's paper click here.
A model of how the two alternative PtdIns(3,4,5)P3-binding and PIF-pocket-dependent mechanisms enable Akt to be efficiently activated and account for resistance to PDK1 inhibitors is shown in the figure above.
A PhD student in Dario Alessi's lab, Ayaz Najafov, characterised the first selective PDK1 inhibitors such as GSK2334470 recently developed by pharmaceutical companies as potential anti-cancer agents.
However, Ayaz found frustratingly that all structurally diverse PDK1 inhibitors he tested were surprisingly ineffective at suppressing Akt activation under conditions that these compounds effectively suppressed activation of other PDK1 substrates such as S6K, RSK and SGK isoforms.
After considerable research Ayaz discovered that resistance to PDK1 inhibitors results from Akt being efficiently recruited to PDK1 via two alternative mechanisms. The first involves ability of Akt and PDK1 to mutually interact with the PI 3-kinase second messenger PtdIns(3,4,5)P3. The second entails recruitment of PDK1 to Akt after its phosphorylation at Ser473 by mTORC2, via a substrate-docking motif termed the PIF-pocket. Ayaz showed that disruption of either the PtdIns(3,4,5)P3 or the Ser473 phosphorylation/PIF-pocket mechanism, only moderately impacts on Akt activation, but induces marked sensitization to PDK1 inhibitors.
The implications of Ayaz's findings is that it will be difficult to ablate Thr308 phosphorylation and hence activation of Akt, employing PDK1 or even mTOR inhibitors as single agents. Ayaz's solution was to combine PDK1 and mTOR inhibitors. Indeed in all cell lines he tested, Ayaz observed that combinations of mTOR and PDK1 inhibitors potently suppresses Thr308 and Ser473 phosphorylation to below-basal levels also inhibiting phosphorylation of Akt substrates. Furthermore, combination of PDK1 and mTOR inhibitors had much greater effects on inhibition of proliferation of all evaluated cancer cell lines, than treatments with either of the inhibitors alone.
Ayaz's results suggests that combination of PDK1 and mTOR inhibitors might represent a new therapeutic strategy for treatment of cancers that harbour mutations elevating Akt activity. To read Ayaz's paper click here.
A model of how the two alternative PtdIns(3,4,5)P3-binding and PIF-pocket-dependent mechanisms enable Akt to be efficiently activated and account for resistance to PDK1 inhibitors is shown in the figure above.