

Background
The DNA in our genomes becomes damaged at a high frequency in all cell types – with tens of thousands of DNA lesions occurring in some cell types in a single day. Left unrectified, such a high level of DNA damage could result in a high level of mutation and genome instability, which would in turn compromise gene expression, cell function and life itself.
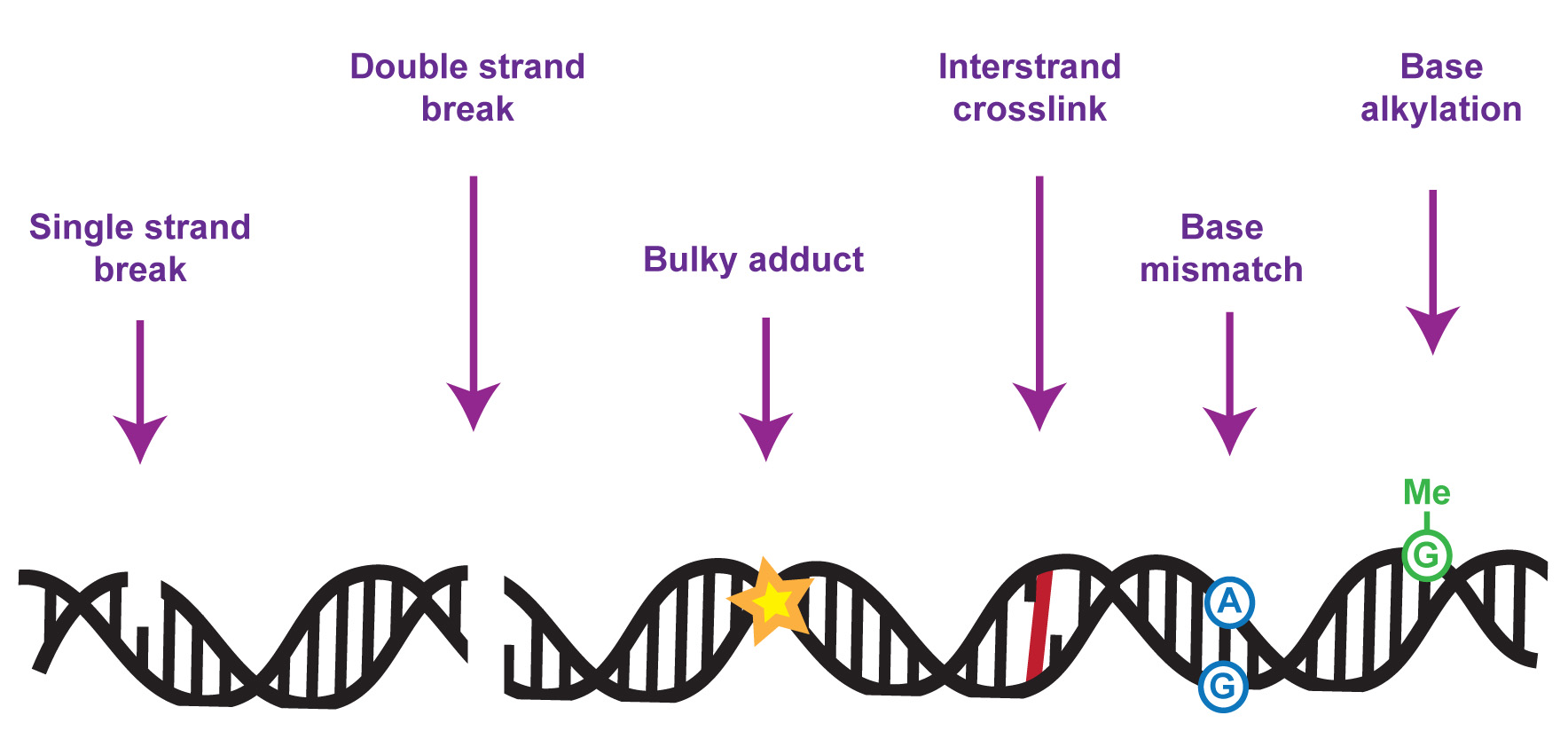
My team focuses on understanding the molecular mechanisms underlying cellular responses to DNA damage, especially to replication or transcription-blocking lesions, with emphasis on control by protein phosphorylation and ubiquitylation. Over the years we have discovered a range of proteins with crucial roles in DNA repair and genome maintenance, and we’ve studied how these proteins work at the molecular level to promote DNA repair. This includes the SLX4 “molecular toolkit” which removes toxic tangles from chromosomes during DNA repair. We also discovered the FAN1 DNA repair nuclease which suppresses chromosome abnormalities by helping to deal with blocked DNA replication forks.
Mutations in genes encoding proteins that control responses to DNA damage cause a wide range of human diseases. Mutations in DNA damage response genes can also cause sterility, cancer, neurological dysfunction, premature aging or death during embryonic development, depending on the pathway involved. So, genome maintenance is vital for human health and well-being. We are keen to understand the mechanisms whereby disease-associated mutations in DNA damage response regulators perturb cell function and cause disease. We are particularly interested in identifying new drug targets and in the “molecular rebalancing” of disease-associated molecular signatures relevant to chromosome maintenance.
A live-imaging platform for identifying new genome maintenance factor
Our work, and that of others, has shown that the timely recruitment of DDR proteins to DNA lesions is crucial for preventing genome instability and human disease. All known DNA damage response (DDR) factors are recruited to sites of genomic DNA damage, and we used this protein property to find new DDR proteins. We have established an automated state-of-the-art cell-based live imaging microscopy platform to enable us to screen protein families for members that are recruited to DNA damage sites. To date, we have screened a number of protein families including the human kinome, and the methodology, cell constructs and cell lines will be made available shortly.
Disease-mutated kinases involved in DNA repair and genome maintenance
CDKL5 – at the nexus of transcription-coupled responses to DNA damage
The first kinase we identified using the imaging platform described above was CDKL5. This is is a poorly understood protein kinase. At the outset, the physiological substrates, cellular roles and activation mechanism of this kinase were not known, and we decided to tackle this problem. We were motivated by that fact CDKL5 is mutated in a rare, debilitating condition known as CDKL5 deficiency disorder (CDD; OMIM 300203; 300672) characterized by epileptic seizures, severe neurodevelopmental delays, impaired motor, language and hand skills and cortical visual impairment. Current treatments focus on management of symptoms, not the underlying cause of the disease. CDD is rare but we now know that CDKL5 is one of the most commonly mutated genes in childhood epilepsy. Thus, the prevalence of CDKL5 mutations is much higher than thought previously.
Early on, we used state-of-the-art phosphoproteomic screening to identify the first physiological targets of CDKL5. The substrates from this first screen were all cytosolic in nature and not obviously linked to DNA damage responses, including the microtubule regulator MAP1S (pSer900) and the centrosome/cilia regulatory factor CEP131 (pSer35). We validated these CDKL5 targets using phospho-specific antibodies, and we identified a CDKL5 consensus sequence: R-P-X-S/T-[A/G/P/S] (the phospho-acceptor residue is underlined) which is a strict prerequisite for substrate phosphorylation. We showed that CDKL5 activity in cells is controlled by autophosphorylation on Tyr171 in the T-loop. We also established cell-based activity assays as well as peptide-based in vitro CDKL5 kinase assays and demonstrated for the first time that CDD-associated mutations severely reduce intrinsic kinase activity. More recently, with our clinical collaborator Helen Leonard we found that the impact of CDKL5 pathogenic mutations on kinase activity correlates with severity of symptoms. Together, our data indicate that it is the loss of CDKL5 target phosphorylation that causes human diseases such as CDD.
What are the nuclear targets of CDKL5? A refined phospho-proteomic screen identified 16 nuclear targets of CDKL5 all phosphorylated on the consensus motif including the EP400 chromatin remodeller, elongin A and TTDN1. Most of the targets are involved in the control of transcription; this provided a vital clue to CDKL5 function in the nucleus, which was a mystery at the time. In this light, we discovered that CDKL5 recruitment to DNA breaks occurs only in actively transcribed regions of the genome and requires nascent mRNA synthesis as well as PAR formation. We demonstrated PAR- and mRNA-dependent phosphorylation of the CDKL5 target elongin A (ELOA) at DNA damage sites, as well as CDKL5 autophosphorylation on the Y171 site required for kinase activity. Most of the nuclear CDKL5 targets, including ELOA, are recruited to DNA damage sites by this same mechanism, thereby juxtaposing kinase and substrates at DNA breaks, facilitating substrate phosphorylation. We showed that CDKL5 kinase activity is required for silencing transcription near DNA breaks, the first definitive nuclear role reported for this kinase.
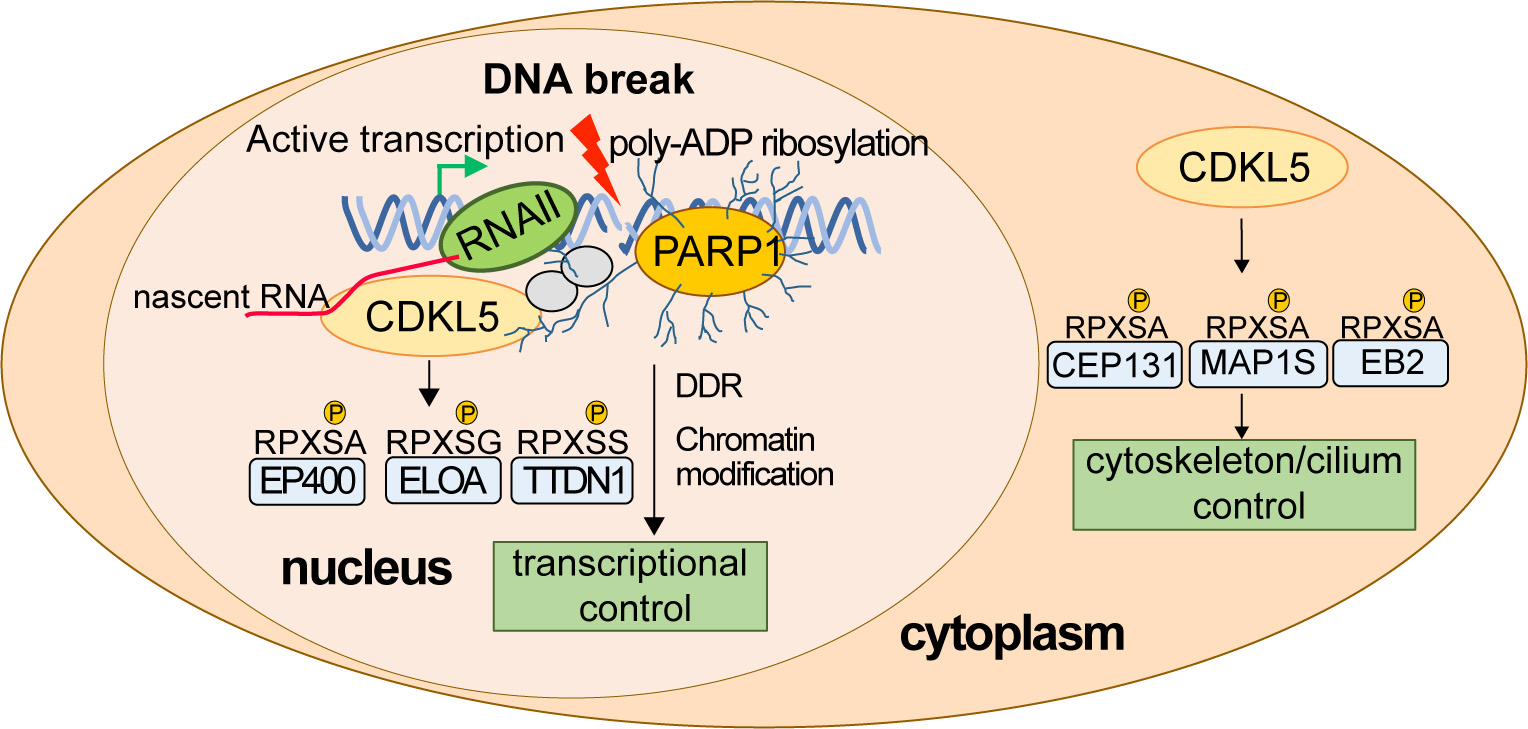
Developing rational therapies to treat the root cause of CDKL5–related diseases requires an understanding of the molecular basis of these diseases and the basic functions of CDKL5. In our lab we are working hard to determine how CDKL5 specifically recognizes DNA breaks at sites of ongoing transcription, how CDKL5 activity is controlled, how CDKL5 recognizes its substrates, how pathological mutations affect CDKL5 activity and function, and which of the functions of CDKL5 is most relevant to human disease. We recently discovered that CDKL5 controls transcriptional elongation in a range of genes even without exogenous DNA damage, and we now wish to build on a body of our unpublished data to study intriguing connections between endogenous DNA breaks, poly ADP ribose, CDKL5 and transcriptional control. We’re also keen to understand how disrupting these mechanisms could lead to diseases such as CDKL5 deficiency disorder (CDD) and epilepsy. We also have plans to carry out “molecular rebalancing” screens to correct the molecular signatures associated with CDKL5 deficiency, which could represent the first rational therapies for treating CDKL5-associated diseases.
NEK1 – a pleiotropic kinase mutated in several distinct disease aetiologies
NEK1 kinase is at once a poorly understood kinase and an attractive drug target. It is amplified in glioma, and depleting NEK1 inhibits the growth of glioma xenografts. Furthermore, NEK1 was reported to control homologous recombination (HR); kinases are druggable targets, and inhibiting NEK1 would inhibit HR which may be a way to treat cancers including HR-defective tumours that switch HR back on after therapy. Although NEK1 has also been implicated in controlling mitosis and primary cilia as well as HR, the underlying mechanisms the substrates were unknown and we decided to apply our expertise to solving this basic problem. Furthermore, NEK1 mutations cause several distinct diseases including amyotrophic lateral sclerosis (ALS), spondylometaphyseal dysplasia (SMD). SMD is a rare skeletal disorder reminiscent of the SPONASTRIME syndrome caused by mutations in TONSL, a HR-promoting histone chaperone identified by our team and others. Therefore, NEK1 mutations may cause SMD by inhibiting HR, but the impact of these mutations on kinase activity or any of the various cellular roles attributed to NEK1 are unknown. We first set out to study the regulation and substrates of NEK1, and at first, we focussed on NEK1 regulation by a potential regulatory subunit, C21ORF2. The poorly characterized gene C21ORF2 is the only gene other than NEK1 mutated in both SMD and ALS. There were hints in the literature that C21ORF2 may interact with so we set out to test if C21ORF2 is a regulatory subunit of NEK1.

We generated antibodies and knockout (KO) cell lines and found that C21ORF2 forms a tight complex with NEK1 in cells. It appears that all of the C21ORF2 in cells is bound to NEK1. We identified a 126 amino acid acidic domain at the C-terminus of NEK1 that is necessary and sufficient for interacting with C21ORF2 termed the CID (C21-interaction domain); Alphafold modelling with Pawel Lis in Dario Alessi’s team revealed that C21ORF2 acts like vice, clamping the NEK1-CID on two interaction surfaces which were validated experimentally. A subset of pathogenic mutations map to the NEK1-CID, and these abolished the interaction with C21ORF2.
Importantly we found that knocking out C21ORF2 (similar to knocking out NEK1) or abolishing the interaction of NEK1 with C21ORF2 disrupts mitosis, ciliogenesis and HR; we went on to show NEK1 kinase activity is important for all these functions. We have recently identified the first physiological substrates of NEK1 throughphospho-proteomic screening (in preparation). We have started from scratch in trying to achieve a detailed understanding of NEK1, in order to answer the following questions: how is NEK1 activity controlled? What are the physiological substrates of NEK1, and which substrates are relevant to the basic cell processes regulated by NEK1? Which cell processes and substrates controlled by NEK1 are most relevant to the different diseases caused by NEK1 mutations? How do pathogenic mutations affect NEK1 function? How does the regulatory subunit of NEK1 we identified impact NEK1 function? We’re taking a multi-disciplinary approach to answer these questions, and to design rational treatments for the “molecular rebalancing” of NEK1-associated diseases.
ATR – a sensor of DNA replication stress
. The ATR protein kinase, which plays critically important roles in the maintenance of genome stability has emerged as a promising anti-cancer drug target. ATR activity becomes even more important in tumour cells harbouring ATM mutations or activated oncogenes such as cyclin E (CCNE1), MYC and RAS than in healthy cells. Berzosertib (Merck) was the first ATR inhibitor (ATRi) to be tested in humans and has entered at least 19 phase I and II clinical trials as a monotherapy in a range of solid tumours. Pharmacodynamic biomarkers are vital for evaluating target engagement in clinical trials, but the standard ATR target pCHK1-Ser345 has proven insufficiently sensitive to monitor ATR activity under basal conditions without co-administration genotoxic drugs such as cisplatin. In collaboration with Merck KGaA, we optimized our phosphoproteomic pipeline to identify ATR biomarkers whose phosphorylation is ultra-sensitive to berzosertib or the structurally unrelated Merck ATRi gartisertib. These analyses generated rich datasets, revealing a wide range of phosphorylation events dependent on ATR, some of which were also dependent on the ATR-activated kinase CHK1.
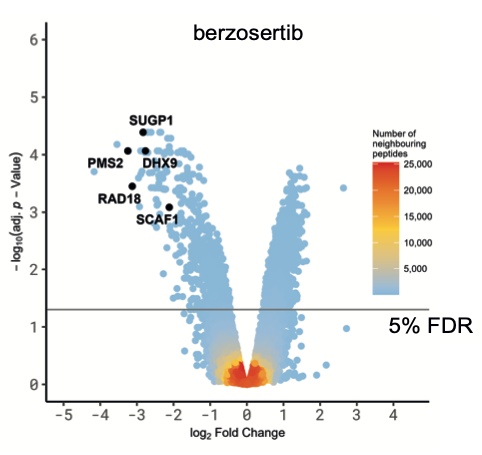
We focussed on the hits common to both ATR inhibitors to rule out off-target effects. Intriguingly, analysis of amino acid sequences surrounding the phosphorylation sites inhibited by ATRi revealed strong enrichment of two different phospho-motifs affected by both inhibitors: the expected pS/pT-Q motif typical of ATR and other PI-(3) kinase-like kinases, and an unexpected motif pSRXXS. The latter motif is likely to be by an ATR-activated kinase(s) which remains to be identified but may correspond to the SRPK-family kinases.
We divided the screening hits common to both ATR inhibitors into three classes: i) ATR targets whose phosphorylation is highly sensitive to ATR inhibition. Some of these proteins – PMS2 and RAD18, for example – are set to become the next generation of ATR biomarkers, which Merck are pursuing; ii) novel ATR targets in known DDR proteins, including conserved sites within functionally annotated domains; these hits include the FANCJ helicase which is phosphorylated on a conserved residue within the helicase domain and the TLK2 kinase which is phosphorylated within the kinase domain by ATR. The data suggest the activity of FANCJ and TLK2 may be controlled by ATR; iii) novel ATR-dependent targets of unknown function. We focussed on the third class of ATR target and subjected them to secondary screening for recruitment to sites of DNA damage. Positives included SCAF1 which is recruited to DNA damage sites in a PARP1- and transcription-dependent manner. We found that the SRI domain of SCAF1 binds to the Ser2/Ser5-phosphorylated form of the RNA polymerase II C terminal repeat domain, and that SCAF1 deletion or depletion reverses the PARP inhibitor sensitivity of BRCA1-KO cells and also restores HR in these cells. Thus, SCAF1 appears to act at the nexus of transcription and genome maintenance.
Chromatin control of genome maintenance
We’re interested in the contribution that DNA packaging into nucleosomes and chromatin makes to genome maintenance. We’re particularly interested in how histone recycling by histone chaperones modulates processes such as DNA repair and transcription. We recently identified a new DNA damage-responsive histone chaperone that is only poorly understood, and we’re using a range of approaches and experimental systems to understand its role in DNA metabolic processes and genome maintenance. In addition, we’re keen to understand the role of chromatin modulating factors in DNA repair processes that occur during S-phase of the cell cycle, and we’re developing new tools and approaches to do so in intact mammalian cells.
Collaboration with the pharmaceutical industry and biotechnology sector
We are also exploring the possibility that inhibiting some of the DNA repair factors we work with might be useful from a therapeutic standpoint in diseases such as cancer. Not only are we developing small molecule inhibitors, but also in developing PROTACs that stimulate degradation of key target proteins in specific disease-relevant genetic backgrounds.
We have major collaborations with Merck KgAA, Pfizer and Beactica Therapeutics. The pharma industry funds the research of five members of the team.
Our values
There are several values that we care about deeply.
Truth: We want to get to the truths behind the questions we’re asking, without bias or attachment to models or pre-conceived notions. We let our data, and only the data, guide us. We’re agnostic about particular outcomes of our experiments, we wish simply to get to the truth.
Robustness: Generating reliable, reproducible data (and top-notch reagents) that stand the test of time is important to us. We place emphasis on using robust methodologies and multiple independent biological replicates to generate data we can rely upon, from which to move forwards. Statistical robustness is important too. Where possible we use blinding in our experiments to help eliminate bias; in addition, members of the team frequently ask others in the team to test if they can reproduce key findings independently before we move forward.
Openness: We believe that we should be pay attention to all observations made in the course of experiments, without bias or prejudice. When addressing a particular question experimentally, we often find we get the answer to a different question to the one we’re asking, which has sometimes turned out to provide breakthroughs! We deposit all of the source data from our papers on repositories such a Mendeley, although now it’s usually possible to publish source data with each individual paper.
Diversity: It really matters to us in the team that we have the fullest possible representation from the various groups that make up our society. It is our belief that the more diverse we are as a group, the greater the diversity of opinions and creativity we’ll have to draw on, which in turns enriches our science and our productivity. We’re a diverse team with multiple ethnicities, religions and nationalities represented, as well as LGBT+ representation. We welcome applications from students and postdocs in our team of all races, religions (or none), gender identifications, sexual orientations, ages, or disability status. We’re a family-friendly lab, open to flexible working for people with children.
Join Us!
The team is looking for talented, motivated, switched-on, diverse, friendly people to join us in addressing key questions about genome maintenance mechanisms relevant to human disease. We have several positions available; informal enquiries can be made by emailing John Rouse (j.rouse@dundee.ac.uk).