Identification of long-sought after substrate of the Parkinson's LRRK2 protein kinase
There was a lot of excitement 11 years ago when it was discovered that autosomal dominant missense mutations within the gene encoding for a previously unstudied protein kinase termed LRRK2 (leucine-rich repeat protein kinase 2) predispose humans to develop Parkinson's disease. Mutations in LRRK2 account for 4% of familial Parkinson's, and are observed in 1% of sporadic PD patients, making it one of the most commonly mutated genes linked to Parkinson's.
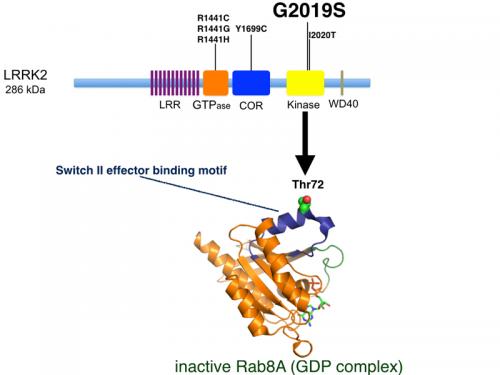
Importantly, the most common Parkinson's- pathogenic mutation is found within the catalytic domain (G2019S) and activates protein kinase activity suggesting inhibitors might offer benefit for the treatment of Parkinson's.
However, a very major stumbling block has been that despite nearly 1500 papers being published to date, no one has this far been able to pinpoint a clear-cut physiological endogenous substrate of LRRK2 that can be validated in independent laboratories. This has greatly hindered our understanding of how LRRK2 is linked to Parkinson's disease as well preclinical evaluation of LRRK2 inhibitors that have been developed by pharmaceutical companies.
In order to identify the key LRRK2 substrates Dario Alessi with generous support from the Michael J Fox Foundation (MJFF) was able to put together an international team of scientists involving researchers in Matthias Mann's laboratory at the Max Planck Institute in Martinsried, the Medical Research Council Protein Phosphorylation and Ubiquitylation Unit at the University of Dundee (MRC PPU), GlaxoSmithKline and Merck in addition to Marco Baptista and Brian Fiske at the MJFF. The key aim of this collaboration was for all teams to work together and make use of each other complementary expertise to exploit state-of-the-art mass spectrometry, genetic and pharmacological approaches to define clear-cut endogenous direct target(s) of LRRK2.
After nearly four years of intensive work our team is delighted to announce that we have at long last identified the first genuine physiological substrate of LRRK2, which comprises a subset of Rab GTPase isoforms including Rab7L1, Rab8A, Rab10 and Rab12. The data indicates LRRK2 phosphorylates these Rab isoforms at a conserved Thr or Ser residue lying in the middle of the key business effector-binding Switch-II motif of Rab GTPases (Thr72 in Rab8A). We show that Parkinson's causing mutations in LRRK2 including the G2019S mutation as well as others such as the R1441G/C (located within the ROC GTPase domain) or Y1699C (located within the COR domain) markedly enhanced LRRK2 phosphorylation of Rab8A and Rab10 isoforms in vivo.
There are ~70 Rab GTPases encoded by the human genome that play central roles in regulating all aspects of membrane dynamics in eukaryotic cells and are master regulators of cargo collection, vesicle formation, vesicle motility, vesicle docking and vesicle fusion. Although 41 Rab isoforms possess a potential LRRK2 phosphorylation site in the middle of the switch II effector binding domain, our initial data indicates that only a subset of these are likely phosphorylated by LRRK2 in cells.
Our results suggest that LRRK2 mediated phosphorylation of Rab isoforms is inhibitory as phosphorylation of Rab isoforms by LRRK2 prevents them from interacting with known effects such a GDIs (required for the insertion of Rab isoforms into membranes) and GDP/GTP exchange factors (e.g. Rabin-8 for Rab8A).
Therefore the key conclusion from our work is that Parkinson's mutations in LRRK2 stimulate the phosphorylation of a subset of Rab isoforms and this inhibits their biological function.
This is an enormously exciting result that we believe is relevant to better understanding Parkinson's as previous work has also linked Rab GTPases to Parkinson's as inherited mutations in two Rab isoforms (Rab7L1/Rab29 and Rab39B) cause Parkinson's–like disease in humans. Furthemore, recent work from MRC-PPU laboratory of Miratul Muqit has found that the PINK1 protein kinase that is also mutated in Parkinson's, indirectly controls the phosphorylation of certain Rab GTPases including Rab8A at a distinct site to LRRK2 (Ser111 on Rab8A). Finally, the work of Susan Lindquist has also linked the binding of alpha-synuclein to Rab8A with Parkinson's disease. Taken together these findings provide mounting evidence that disruption on Rab GTPase biology could at the heart of better understanding and treating Parkinson's disease.
We anticipate that our work on elucidating the LRRK2-Rab signalling network could open up a new era of research which could lead to improved opportunities for better understanding LRRK2 biology and how mutations cause Parkinson's. Reagents we have elaborated to monitor LRRK2 phosphorylation of Rab GTPases could aide pharmaceutical companies advance LRRK2 inhibitors into the clinic as it should now become possible to readily assess the impact that LRRK2 inhibitors have by assessing the level of LRRK2 phosphorylated Rabs in vivo. It would be fascinating to explore whether monitoring Rab phosphorylation could also be used as a diagnostic method to assess whether Parkinson's patients would benefit from drugs that targeted LRRK2. Finally tools to study Rab phosphorylation will help address the question of whether Rab GTPases are hyper-phosphorylated in Parkinson's patients that do not have LRRK2 mutations and whether such patients would benefit from LRRK2 inhibitors.
Read the full article here: http://elifesciences.org/content/early/2016/01/28/eLife.12813
We have also initiated a search for two postdoctoral researchers to work in this area for further information please contact Dario Alessi (d.r.alessi@Dundee.ac.uk) or click here.
Importantly, the most common Parkinson's- pathogenic mutation is found within the catalytic domain (G2019S) and activates protein kinase activity suggesting inhibitors might offer benefit for the treatment of Parkinson's.
However, a very major stumbling block has been that despite nearly 1500 papers being published to date, no one has this far been able to pinpoint a clear-cut physiological endogenous substrate of LRRK2 that can be validated in independent laboratories. This has greatly hindered our understanding of how LRRK2 is linked to Parkinson's disease as well preclinical evaluation of LRRK2 inhibitors that have been developed by pharmaceutical companies.
In order to identify the key LRRK2 substrates Dario Alessi with generous support from the Michael J Fox Foundation (MJFF) was able to put together an international team of scientists involving researchers in Matthias Mann's laboratory at the Max Planck Institute in Martinsried, the Medical Research Council Protein Phosphorylation and Ubiquitylation Unit at the University of Dundee (MRC PPU), GlaxoSmithKline and Merck in addition to Marco Baptista and Brian Fiske at the MJFF. The key aim of this collaboration was for all teams to work together and make use of each other complementary expertise to exploit state-of-the-art mass spectrometry, genetic and pharmacological approaches to define clear-cut endogenous direct target(s) of LRRK2.
After nearly four years of intensive work our team is delighted to announce that we have at long last identified the first genuine physiological substrate of LRRK2, which comprises a subset of Rab GTPase isoforms including Rab7L1, Rab8A, Rab10 and Rab12. The data indicates LRRK2 phosphorylates these Rab isoforms at a conserved Thr or Ser residue lying in the middle of the key business effector-binding Switch-II motif of Rab GTPases (Thr72 in Rab8A). We show that Parkinson's causing mutations in LRRK2 including the G2019S mutation as well as others such as the R1441G/C (located within the ROC GTPase domain) or Y1699C (located within the COR domain) markedly enhanced LRRK2 phosphorylation of Rab8A and Rab10 isoforms in vivo.
There are ~70 Rab GTPases encoded by the human genome that play central roles in regulating all aspects of membrane dynamics in eukaryotic cells and are master regulators of cargo collection, vesicle formation, vesicle motility, vesicle docking and vesicle fusion. Although 41 Rab isoforms possess a potential LRRK2 phosphorylation site in the middle of the switch II effector binding domain, our initial data indicates that only a subset of these are likely phosphorylated by LRRK2 in cells.
Our results suggest that LRRK2 mediated phosphorylation of Rab isoforms is inhibitory as phosphorylation of Rab isoforms by LRRK2 prevents them from interacting with known effects such a GDIs (required for the insertion of Rab isoforms into membranes) and GDP/GTP exchange factors (e.g. Rabin-8 for Rab8A).
Therefore the key conclusion from our work is that Parkinson's mutations in LRRK2 stimulate the phosphorylation of a subset of Rab isoforms and this inhibits their biological function.
This is an enormously exciting result that we believe is relevant to better understanding Parkinson's as previous work has also linked Rab GTPases to Parkinson's as inherited mutations in two Rab isoforms (Rab7L1/Rab29 and Rab39B) cause Parkinson's–like disease in humans. Furthemore, recent work from MRC-PPU laboratory of Miratul Muqit has found that the PINK1 protein kinase that is also mutated in Parkinson's, indirectly controls the phosphorylation of certain Rab GTPases including Rab8A at a distinct site to LRRK2 (Ser111 on Rab8A). Finally, the work of Susan Lindquist has also linked the binding of alpha-synuclein to Rab8A with Parkinson's disease. Taken together these findings provide mounting evidence that disruption on Rab GTPase biology could at the heart of better understanding and treating Parkinson's disease.
We anticipate that our work on elucidating the LRRK2-Rab signalling network could open up a new era of research which could lead to improved opportunities for better understanding LRRK2 biology and how mutations cause Parkinson's. Reagents we have elaborated to monitor LRRK2 phosphorylation of Rab GTPases could aide pharmaceutical companies advance LRRK2 inhibitors into the clinic as it should now become possible to readily assess the impact that LRRK2 inhibitors have by assessing the level of LRRK2 phosphorylated Rabs in vivo. It would be fascinating to explore whether monitoring Rab phosphorylation could also be used as a diagnostic method to assess whether Parkinson's patients would benefit from drugs that targeted LRRK2. Finally tools to study Rab phosphorylation will help address the question of whether Rab GTPases are hyper-phosphorylated in Parkinson's patients that do not have LRRK2 mutations and whether such patients would benefit from LRRK2 inhibitors.
Read the full article here: http://elifesciences.org/content/early/2016/01/28/eLife.12813
We have also initiated a search for two postdoctoral researchers to work in this area for further information please contact Dario Alessi (d.r.alessi@Dundee.ac.uk) or click here.