Mutations that increase the kinase activity of LRRK2 cause Parkinson's disease and therapies that reduce LRRK2 kinase activity are being tested in clinical trials.
Numerous rare variants that alter the amino acid sequence of LRRK2 have been reported that are of unknown clinical significance. Alexia Kalogeropulou, Elena Purltye and Francesca Tonelli with support from Esther Sammler and Shalini Padmanabhan from The Michael J Fox Foundation were keen to establish a workflow to define whether a given variant impacts LRRK2 kinase activity.
We initially selected 100 LRRK2 variants that had been linked in at least one manuscript to Parkinson’s. The vast majority of these had not been previously investigated at the cellular level.
To get the project off the ground a tremendous amount of work was undertaken by Melanie Wightman to clone all these LRRK2 variants in the same mammalian expression vector.
We then expressed each of the variants in HEK293 cells and assessed kinase activity by monitoring LRRK2 mediated phosphorylation Rab10 and Rab12 and comparing activity with wild type LRRK2. Variants that were deemed to increase activity in the primary screen were reanalysed in a secondary screen.
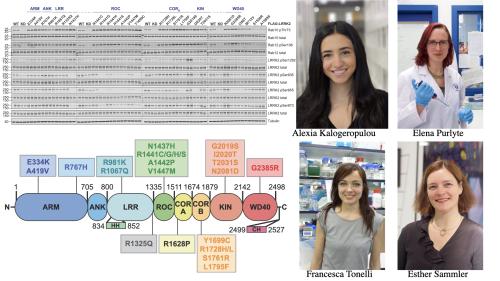
This work revealed 23 variants the majority of which had not been previously reported that were observed to robustly stimulate LRRK2 kinase activity at least 1.5-fold in the HEK293 cell assay. These variants are located within the N-terminal non-catalytic regions [ARM (E334K, A419V), ANK(R767H), LRR (R1067Q, R1325Q)], as well as variants predicted to destabilise the ROC:CORB interface [ROC (A1442P, V1447M), CORA (R1628P) CORB (S1761R, L1795F)] and COR:COR dimer interface [CORB (R1728H/L)] (see Figure left hand panel).
We also found that most activating variants decrease LRRK2 biomarker site phosphorylation (pSer935/pSer955/pSer973), consistent with the notion of previous work that the active/ pathogenic kinase conformation reduces their phosphorylation.
Our studies also suggest that the impact of variants on kinase activity is best evaluated by deploying a HEK293 cell overexpression assay, compared to a biochemical kinase assay; only a select few activating LRRK2 variants from the cell overexpression assay [CORB (Y1699C, R1728H/L, S1761R) and kinase (G2019S, I2020T, T2031S)], also enhanced kinase activity of LRRK2 in and in vitro assay. The reasons for this are not clear but emphasize that other factors in cells such as membrane localization are likely to influence kinase activity.
Much previous work by other groups has demonstrated that the presence of a Type I kinase inhibitor, promotes the binding of overexpressed LRRK2 to microtubules. This prompted us to investigate how the variants impacted microtubule binding which revealed that 12 variants including several that activate LRRK2 and have been clinically linked to Parkinson’s, suppressed microtubule association in the presence of a Type I kinase inhibitor. These included the LRRK2 variants [ARM(M712V), LRR(R1320S), ROC (A1442P, K1468E, S1508R), CORA(A1589S), CORB (Y1699C, R1728H/L) and WD40(R2143M, S2350I, G2385R)].
There is still much work to do as LRRK2 is a large protein with 2527 residues and over 1000 variants have been reported thus far. Many people with PD will likely carry additional rare LRRK2 variants not tested in this and other studies. With DNA sequencing becoming more readily available and LRRK2 kinase inhibitors entering clinical trials, a concrete set of functional parameters and experimental workflows will need to be available to help clinicians assess whether any given LRRK2 variant is likely to increase kinase activity.
We advocate the use of the HEK293 cell overexpression assay as the gold standard approach to assess whether a variant enhances LRRK2 activity. We also discuss that work undertaken by Andrew West’s group in calculating a REVEL pathogenic algorithm score together with an conservation evolutionary score for each variant is also useful for assessing pathogenic mutations. We find that the majority of all the activating variants also display a high REVEL and/or evolutionary conservation score.
In ongoing work, Esther Sammler’s laboratory is experimentally assessing additional LRRK2 variants linked to Parkinson’s, and we plan to continue assessing new variants as these are reported. In the future our goal is to deposit all experimental data, REVEL and evolutionary conservation scores for each variant into a publicly accessible database as soon as the data become available.
It is our hope that this information will aide clinicians in interpreting LRRK2 variants in terms of their pathogenicity for genetic counselling. Our biochemical analysis would help stratify people harbouring LRRK2 variants and would assist in identifying individuals with activating mutations for LRRK2-targeting clinical trials, as well as stimulate further mechanistic and structural analysis to better understand how these mutations enhance LRRK2 kinase activity.
We believe that similar functional workflows could be used to catalogue the impact that other PD-associated genes, including the lysosomal enzyme glucocerebrosidase (GBA1), of which there are numerous variants of unknown clinical function.
We also thank the contributions of Sven Lange who undertook detailed structural analysis of the variants to understand the structural impact that these have, as well as Alan Prescott for aiding with microscopy studies.
This study was supported the joint efforts of The Michael J. Fox Foundation for Parkinson’s Research (MJFF) and Aligning Science Across Parkinson’s (ASAP) initiative. MJFF administers the grant (ASAP-000463-Alessi lab) on behalf of ASAP and itself. The D.R.A lab is also supported by the UK Medical Research Council [grant number MC_UU_00018/1] and the pharmaceutical companies supporting the Division of Signal Transduction Therapy Unit.
To read a copy of our paper click here