MRC-PPU research uncovers exciting new role for protein ubiquitylation in regulating blood pressure
Gordon's hypertension syndrome is caused by mutations that increase expression of WNK1 protein kinase as well as specific missense mutations lying within a non-catalytic region of the WNK4 protein kinase. Patients with this condition suffer from high blood pressure and hyperkalemia (high serum potassium), and can be treated using thiazide diuretic hypertension drugs that inhibit the NCC ion co-transporter in the kidney. Much research from our Unit and elsewhere points towards WNK1/WNK4 kinases controlling blood pressure by activating two related kinases termed-SPAK and OSR1 that once switched on regulate hypertension blood pressure by modulating the activity of ion co-transporters in the kidney termed NCC and NKCC2.
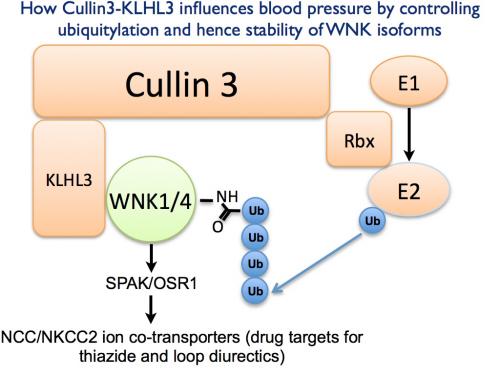
Exciting recent studies by the laboratories of Richard Lifton at Yale and Xavier Jeunemaitre in Paris have identified new players that control Gordon's syndrome. These researchers identified over 50 patients, in which Gordon's syndrome was caused by mutations in Ubiquitin E3 ligase components termed Cullin-3 (CUL3) or Kelch-like 3 (KLHL3) rather than WNK isoforms.
Previous work suggested that CUL3 and KLHL3 would form a heterodimeric complex. Data indicated that CUL3 would function as the catalytic entity mediating the ubiquitylation of substrates whereas KLHL3 would operate as the substrate recognition moiety directing the E3 ligase complex to its specific cellular substrates.
To explore how a CUL3:KLHL3 complex might operate to control blood pressure a Postdoc in Dario Alessi's lab, Akihito Ohta, immunoprecipitated KLHL3 and strikingly found that it associated strongly with WNK1, WNK2 and WNK3 isoforms as well as CUL3. However, in parallel studies no interaction of KLHL3 with other components of the WNK signalling pathway such as SPAK/OSR1 or NCC/NKCC1 was observed. Interestingly, many dominant KLHL3 disease mutations analysed inhibited binding to either WNK1 or CUL3, indicating that the association of WNK isoforms with KLHL3 is relevant to Gordon's syndrome.
A postdoc in Thimo Kurz's lab, Frances-Rose Schumacher, then got together with Axel Knebel and Clare Johnson in the MRC-PPU ubiquitylation component purification and assay team, to generate recombinant wild type and a non-WNK1 binding disease mutant CUL3:KLHL3 complex. Frances-Rose was then able to show that wild type but not the disease mutant complex potently ubiquitylated WNK1 in vitro. Consistent with CUL3 regulating ubiquitylation and stability of WNK1, Akihito was next able to demonstrate that siRNA-mediated knockdown of CUL3 moderately increased WNK1 protein levels and kinase activity in HeLa cells.
Akihito then mapped the KLHL3 interaction site in to a non-catalytic moiety located just C-terminal to the kinase catalytic region of WNK1 (residues 479 to 667). Interestingly, the equivalent region in WNK4 encompasses residues that are mutated in Gordon syndrome patients. Strikingly, Akihitio found that the Gordon's disease causing WNK4[E562K] and WNK4[Q565E] mutations as well as the equivalent mutation in WNK1[479-667] fragment, abolished ability to interact with KLHL3.
These results suggest that mutations in WNK4 that cause hypertension exert their effects by hindering the interaction with KLHL3:CUL3. More work is required to establish this concept, but our prediction is that these missense mutations in WNK4 exert their physiological effects by ablating KLHL3 binding thereby leading to reduced ubiquitylation and hence enhanced expression of WNK4. If this was the case it could result in inappropriate activation of the SPAK/OSR1 kinases resulting in overstimulation of the NCC/NKCC2 ion co-transporters. This would lead to too much salt retention and hence hypertension. In future work it would be critical to properly compare expression levels of WNK4 in WNK4[D561A] knock-in mice that have been generated by the Uchida laboratory in Japan or even in Gordon's syndrome patients.
All our results points towards Gordon's syndrome causing mutations in WNK1, WNK4, KLHL3 and probably CUL3 leading to hypertension by inducing the overexpression of WNK isoforms. Our data suggests that the CUL3-KLHL3 E3 ligase complex regulates blood pressure via its ability to interact with and ubiquitylate WNK isoforms. More generally this research reveals how mutations that disrupt the ability of an E3 ligase to interact and ubiquitylate a critical cellular substrate such as WNK isoforms can trigger a chronic disease such as hypertension.
To read Akihito and Frances-Rose's paper describing these results click here.
Exciting recent studies by the laboratories of Richard Lifton at Yale and Xavier Jeunemaitre in Paris have identified new players that control Gordon's syndrome. These researchers identified over 50 patients, in which Gordon's syndrome was caused by mutations in Ubiquitin E3 ligase components termed Cullin-3 (CUL3) or Kelch-like 3 (KLHL3) rather than WNK isoforms.
Previous work suggested that CUL3 and KLHL3 would form a heterodimeric complex. Data indicated that CUL3 would function as the catalytic entity mediating the ubiquitylation of substrates whereas KLHL3 would operate as the substrate recognition moiety directing the E3 ligase complex to its specific cellular substrates.
To explore how a CUL3:KLHL3 complex might operate to control blood pressure a Postdoc in Dario Alessi's lab, Akihito Ohta, immunoprecipitated KLHL3 and strikingly found that it associated strongly with WNK1, WNK2 and WNK3 isoforms as well as CUL3. However, in parallel studies no interaction of KLHL3 with other components of the WNK signalling pathway such as SPAK/OSR1 or NCC/NKCC1 was observed. Interestingly, many dominant KLHL3 disease mutations analysed inhibited binding to either WNK1 or CUL3, indicating that the association of WNK isoforms with KLHL3 is relevant to Gordon's syndrome.
A postdoc in Thimo Kurz's lab, Frances-Rose Schumacher, then got together with Axel Knebel and Clare Johnson in the MRC-PPU ubiquitylation component purification and assay team, to generate recombinant wild type and a non-WNK1 binding disease mutant CUL3:KLHL3 complex. Frances-Rose was then able to show that wild type but not the disease mutant complex potently ubiquitylated WNK1 in vitro. Consistent with CUL3 regulating ubiquitylation and stability of WNK1, Akihito was next able to demonstrate that siRNA-mediated knockdown of CUL3 moderately increased WNK1 protein levels and kinase activity in HeLa cells.
Akihito then mapped the KLHL3 interaction site in to a non-catalytic moiety located just C-terminal to the kinase catalytic region of WNK1 (residues 479 to 667). Interestingly, the equivalent region in WNK4 encompasses residues that are mutated in Gordon syndrome patients. Strikingly, Akihitio found that the Gordon's disease causing WNK4[E562K] and WNK4[Q565E] mutations as well as the equivalent mutation in WNK1[479-667] fragment, abolished ability to interact with KLHL3.
These results suggest that mutations in WNK4 that cause hypertension exert their effects by hindering the interaction with KLHL3:CUL3. More work is required to establish this concept, but our prediction is that these missense mutations in WNK4 exert their physiological effects by ablating KLHL3 binding thereby leading to reduced ubiquitylation and hence enhanced expression of WNK4. If this was the case it could result in inappropriate activation of the SPAK/OSR1 kinases resulting in overstimulation of the NCC/NKCC2 ion co-transporters. This would lead to too much salt retention and hence hypertension. In future work it would be critical to properly compare expression levels of WNK4 in WNK4[D561A] knock-in mice that have been generated by the Uchida laboratory in Japan or even in Gordon's syndrome patients.
All our results points towards Gordon's syndrome causing mutations in WNK1, WNK4, KLHL3 and probably CUL3 leading to hypertension by inducing the overexpression of WNK isoforms. Our data suggests that the CUL3-KLHL3 E3 ligase complex regulates blood pressure via its ability to interact with and ubiquitylate WNK isoforms. More generally this research reveals how mutations that disrupt the ability of an E3 ligase to interact and ubiquitylate a critical cellular substrate such as WNK isoforms can trigger a chronic disease such as hypertension.
To read Akihito and Frances-Rose's paper describing these results click here.