Much research has focused on understanding how mutations in a protein kinase termed LRRK2, predisposes to Parkinson’s disease. Most pathogenic mutations occur within the kinase domain (G2019S) and GTPase ROC/COR domain (R1441G and Y1699C) of LRRK2.
Recent studies undertaken in the Alessi, Mann and Pfeffer labs, have revealed that LRRK2 phosphorylates a group of Rab GTPase proteins including Rab29 (also known as Rab7L1), within the effector‐binding switch II motif. This enables Rab proteins to interact with new effectors including RILPL1 and RILPL2.
Mutations located within the kinase domain such as G2019S, directly stimulate protein kinase activity of LRRK2.
Although the R1441G and Y1699C ROC/COR domain mutations enhance GTP binding as well as Rab protein phosphorylation in cells, they do not stimulate LRRK2 kinase activity in vitro.
Previous work indicated that Rab29, located within the PARK16 locus mutated in Parkinson's patients, operates in a common pathway with LRRK2. How Rab29 controlled LRRK2 was not known.
Francesca Tonelli (Postdoc in the Alessi lab) set out to uncover how Rab29 might control LRRK2. She found that Rab29 potently activated LRRK2 in overexpression experiments. Excitingly, Francesca noticed that the R1441G and Y1699C ROC/COR mutants were activated by Rab29 to a much greater extent than wild type or G2019S LRRK2.
Our collaborators Herschel Shrikant and Suzanne Pfeffer at the University of Stanford obtained stunning data, that revealed Rab29 recruits LRRK2 to the trans‐Golgi network where it stimulates LRRK2 activity. Consistent with Francesca’s activation date, pathogenic R1441G/C and Y1699C mutants, were also more readily recruited to the Golgi than wild type LRRK2.
Rab29 activated LRRK2, was also found to cause significant fragmentation of the Golgi, in a manner that was largely reversed with an LRRK2 inhibitor.
Careful comparison of the mechanism by which another Rab protein (Rab32) binds to one of its known Ankyrin-domain effectors (VARP), enabled Elena Purlyte (Alessi lab PhD student) to identify a series of highly conserved Leu residues within the LRRK2 ankyrin domain required for Rab29‐mediated Golgi recruitment and kinase activation.
Consistent with Rab29 activating LRRK2, Adil Sarhan (Alessi lab postdoc) found that knockout of Rab29 in A549 cells reduced endogenous LRRK2‐mediated phosphorylation of Rab10.
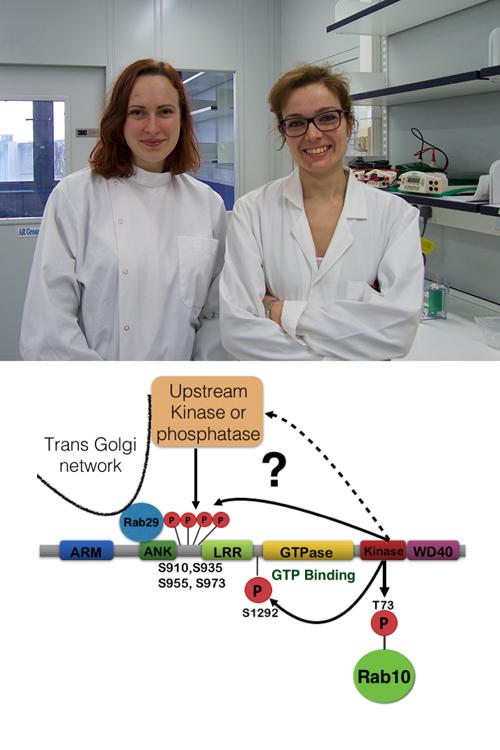
Elena also unexpectedly found that mutations within the LRRK2 Ankyrin domain that prevented LRRK2 from interacting with Rab29, strikingly inhibited phosphorylation of a cluster of highly studied biomarker sites (Ser910, Ser935, Ser955 and Ser973).
Mutations that block GTP binding to the ROC/Cor GTPase domain of LRRK2 also blocked biomarker phosphorylation. Herscel also showed that GTP non-binding mutant of LRRK2 could not be recruited by Rab29 to the Golgi.
Francesca also obtained data suggesting that LRRK2 mediated phosphorylation of Rab29 at Ser71 and Thr72 may act as a feedback loop mechanism, dissociating interaction of LRRK2 with Rab29. This could represent a mechanism by which activated LRRK2 is released from the Golgi once it is activated by Rab29.
Overall, the data suggest that Rab29 is a master regulator of LRRK2, controlling its activation, localization, and potentially biomarker phosphorylation. Mutations that promote GTP binding to LRRK2 such as R1441G likely induce a conformational change that promotes recruitment and activation by Rab29.
If inhibitors that block Rab29 binding and activation of LRRK2 could be developed, these would be expected to suppress LRRK2 activity, and perhaps offer therapeutic potential for the treatment of Parkinson's disease.
In future work, it will be critical to delineate the mechanism by which Rab29 binding and recruitment to the Golgi membrane activates LRRK2. It will also be important to decipher how Rab29 promotes phosphorylation of LRRK2 biomarker phosphorylation sites (S910, S935, S955, S973), and whether this is mediated through an autophosphorylation mechanism or by a distinct upstream proteins kinase.
To read a copy of our paper on the control of LRRK2 by Rab29 click here http://emboj.embopress.org/content/early/2017/12/06/embj.201798099