Google Scholar | PubMed | Biography
How Does Dysregulated Signal Transduction Cause Intellectual Disability?

Intellectual disability is a major mental health problem that affects 1-3% of the world population and is characterised by varying clinical features including impaired intellectual development, autism, facial dysmorphism and microcephaly. Surprisingly, genetic studies have shown that intellectual disability is frequently caused by mutations in genes that encode poorly-studied signalling enzymes. Therefore, the goal of our lab is to understand how protein phosphorylation and ubiquitylation is disrupted in intellectual disability patients using pluripotent stem cells as a model. In our research, we aim to employ our expertise in signal transduction to unravel cell signalling pathways that go awry in intellectual disability patients, with the ultimate objective of identifying potential therapeutic targets.
Our work over the past 5 years has uncovered a novel pathway that represents a paradigm for how dysregulated signalling causes intellectual disability. This began with a landmark study reporting how variants in the E3 ubiquitin ligase RNF12 (also known as RLIM) specifically disrupt ubiquitin signalling and neuronal differentiation to cause a syndromic form of intellectual disability named Tonne-Kalscheuer syndrome (TOKAS). This provided the first insights into how dysregulated RNF12 signalling leads to intellectual disability. We have since led efforts to characterise novel RNF12 intellectual disability variants in collaboration with clinical colleagues and made recent progress in unravelling further RNF12 functions and regulatory mechanisms. Importantly, we discovered that Ser-Arg Splicing Factor Protein Kinase family (SRPK1-3) phosphorylates and activates RNF12, and that the SRPK-RNF12 pathway controls expression of neurodevelopmental genes, a function that is disrupted in intellectual disability. We have since collaborated with clinical groups to identify loss-of-function SRPK3 gene variants in intellectual disability families and are working with MRC-PPU colleague Satpal Virdee to employ activity-based E3 ligase probes in monitoring dysregulated E3 ligase function in intellectual disability patients. In complementary work, we uncovered a further SRPK-RNF12-dependent developmental pathway involved in gametogenesis that is disrupted in TOKAS patients.
Previously, we have used cutting-edge screening technologies to uncover several unappreciated signalling pathways that regulate embryonic stem cell pluripotency and differentiation, opening new avenues of research in the field. Highlights include a chemical-genetic screen that identified a new function for the poorly studied MAP kinase ERK5 in embryonic stem cells during the previous quinquennium. We have since characterised novel mechanisms by which ERK5 regulates the poorly understood process of stem cell rejuvenation. We also developed a quantitative phosphoproteomic approach, which uncovered a new function for EPH receptors in pluripotent cells to reveal how cellular differentiation and organisation are coordinated during development.
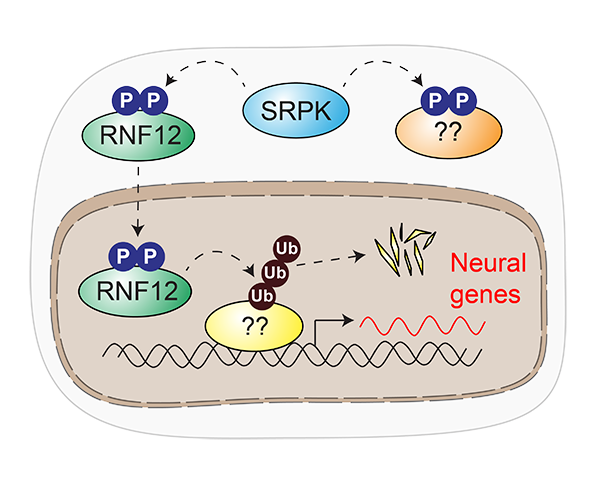
Our research has uncovered a paradigm for how dysregulated signal transduction causes intellectual disability. SRPK phosphorylates RNF12, which stimulates RNF12 catalytic activity and promotes nuclear anchoring. RNF12 in turn drives ubiquitylation of nuclear transcriptional regulators to modulate neurodevelopmental gene expression. Our work also proposes that further SRPK-dependent pathways are relevant for intellectual disability.
Current Projects
Our research aims to elucidate molecular and cellular principles underpinning intellectual disability, by using our expertise in signal transduction to determine how signalling is disrupted in patients. We will employ biochemistry, chemical genetics, transcriptomics, proteomics and genome editing in human pluripotent stem cell-derived neurons and a mouse model to unravel mechanisms and functions of the SRPK-RNF12 pathway. This work will uncover fundamental molecular, cellular and neurological mechanisms that are disrupted in patients, elucidate the wider landscape of intellectual disability signalling and ultimately illuminate new therapeutic strategies. Our work is highly collaborative, involving several teams within the MRC-PPU, external collaborators including Dr Sara Wells at the MRC Mary Lyon Centre (Harwell), and expert developmental neurobiologist Prof Kate Storey and the Human Pluripotent Stem Cell Facility (both University of Dundee).
What are the key molecular targets of SRPK and RNF12 that are relevant for intellectual disability?
A core objective is to identify substrates and gene expression programmes that are dysregulated following SRPK-RNF12 pathway disruption. We will use human induced pluripotent stem cell (hiPSC)-derived neurons and selectively inhibit SRPK-RNF12 signalling by CRISPR/Cas9 engineering a RNF12/RLIM TOKAS intellectual disability variant (R599C) and by employing an inhibitor-resistant SRPK2 allele that we recently developed. We will then deploy quantitative proteomics and transcriptomics to identify substrates and transcriptional signatures that are dysregulated following SRPK-RNF12 pathway inhibition. This will illuminate how dysregulated SRPK-RNF12 signalling disrupts key molecular processes that underpin intellectual disability.
Excitingly, an initial phosphoproteomic exploration of the wider SRPK signalling network suggests that SRPK phosphorylates other proteins implicated in intellectual disability. Therefore, it is our goal to generate a comprehensive map of SRPK substrates and neurodevelopmental functions by developing a quantitative phosphoproteomics pipeline in hiPSC-derived neurons. Collaboration with Prof Andrew Crosby (University of Exeter), an expert in clinical genetics of intellectual disability, will enable us to pinpoint and prioritise SRPK substrates encoded by high-confidence causative intellectual disability genes. SRPK phosphorylation sites will be mapped and monitored in hiPSC-derived neurons, and we will define the role of SRPK substrate phosphorylation in regulation of functional parameters such as activity, localisation and stability. We will then investigate SRPK substrate functions during human neural development and explore how they are disrupted by intellectual disability variants.
A priority SRPK substrate identified in our initial phosphoproteomic screens is Fragile X-syndrome Related protein (FXR2), a member of the FXR family of mRNA binding proteins that includes FXR1 and FMR1. FXR proteins associate with ribosomes to control localised translation in neurons. FMR1 is mutated in Fragile-X syndrome intellectual disability, whilst FXR2 is also required for correct neuronal functioning. Emerging evidence suggests that FXR proteins are regulated by phosphorylation, which drives a phase transition to form Fragile X granules and modulate mRNA translation. Current focus is on investigating the function of SRPK signalling to FXR proteins during hiPSC neural differentiation. We will explore Fragile-X granule formation and translational regulation in hiPSC-derived cortical neural progenitors, deploying quantitative proteomics and transcriptomics to identify FXR2 translational targets. This will serve as a starting point to address the role of SRPK in regulating localised protein translation in hiPSC-derived cortical progenitors and neurons.
What is the mechanism by which SRPK activates RNF12?
An exciting mechanistic question we are seeking to address is how SRPK phosphorylation regulates RNF12 catalytic activity. SRPK phosphorylates RNF12 in a region distal to the catalytic RING domain, suggesting that the SRPK-RNF12 pathway employs a novel E3 ubiquitin ligase activation mechanism. We aim to provide molecular insight into the SRPK-dependent RNF12 activation using cryo-EM. We will continue an established collaboration with former MRC-PPU PI, Prof Helen Walden (University of Glasgow), who is an expert in employing cryo-EM to make fundamental insights into E3 ubiquitin ligase regulation. Affinity purification mass-spectrometry has identified 14-3-3σ/Sfn as a major interaction partner that is recruited to RNF12 following phosphorylation by SRPK, suggesting that 14-3-3σ plays a key role in RNF12 regulation. Therefore, we will investigate the impact of 14-3-3σ recruitment to SRPK phosphorylated RNF12 on structure and catalytic regulation. This will pinpoint in molecular detail the mechanisms by which SRPK phosphorylation promotes RNF12 substrate ubiquitylation, which we will verify using in vitro and cell-based assays. We will also identify mechanisms by which RNF12 TOKAS variants impact on catalytic activity. We anticipate this will provide a detailed molecular understanding of a key molecular switch governing the SRPK-RNF12 axis functions, and how it is dysregulated in intellectual disability.
Which neurodevelopmental functions of SRPK-RNF12 signalling are disrupted in intellectual disability patients?
SRPK-RNF12 signalling controls neural genes and differentiation, suggesting that disrupted neurodevelopment may underpin intellectual disability in patients. Therefore, we aim to investigate the regulation and function of SRPK-RNF12 in human models of neural development. We will determine when and where the SRPK-RNF12 pathway is active during hiPSC neuronal differentiation, employing RNF12 phosphorylation as a biomarker. In collaboration with Prof. Kate Storey, we will define the function of SRPK-RNF12 signalling during human neural development by deploying inhibitor-resistant SRPK and RNF12/RLIM TOKAS hiPSC lines in hiPSC-derived cortical neural rosettes, which is a sophisticated in vitro model of human cortical neurogenesis.
To address the impact of SRPK-RNF12 pathway disruption in the intact nervous system, we have developed the first mouse model of dysregulated SRPK-RNF12 signalling in collaboration with Dr Sara Wells (MRC Mary Lyon Centre, Harwell). This mouse harbours an RNF12 R575C knock-in mutation, equivalent to the human RNF12 R599C TOKAS intellectual disability variant, with impaired E3 ubiquitin ligase activity. We will define brain regions in which the SRPK-RNF12 pathway is active, then dissect and harvest brain tissue to determine whether SRPK-RNF12 substrates and gene expression signatures are dysregulated in the intact nervous system. We will also determine the impact of SRPK-RNF12 pathway dysregulation on neurological function, employing established behavioural analysis platforms to investigate social behaviours. If interesting behavioural observations suggest neurological dysfunction, we will collaborate with expert neurobiologists for follow up studies into underlying neurological mechanisms.
An overarching goal of this project is to investigate whether dysregulated SRPK-RNF12 signalling is a hallmark of intellectual disability. To address this, we are curating a panel of hiPSC lines harbouring patient-derived intellectual disability variants in signalling components, including novel components of the SRPK-RNF12 signalling network. We will differentiate cortical neural progenitors to investigate how these components integrate with the SRPK-RNF12 pathway. This ambitious approach will enable us to pursue our long-term objective of comprehensively mapping the SRPK-RNF12 signalling network and interrogating the wider role of signal dysregulation in intellectual disability.
Diversity
It matters to us that we have the fullest possible representation of the various groups that make up our society. It is our belief that the more diverse we are, the greater the diversity of opinions and creativity we’ll have to draw on, which in turns enriches our science. We’re a diverse team with multiple ethnicities, religions and nationalities represented. We welcome applications from students and postdocs in our team of all races, beliefs, gender identification, sexual orientation, age, or disability status. We’re a family-friendly lab, open to flexible working for people with children.
Join Us!
The team is looking for talented, motivated, switched-on, diverse, friendly people to join us. We have several positions available; informal enquiries can be made by emailing Greg Findlay (g.m.findlay@dundee.ac.uk).
