New role for the hypertension WNK-regulated SPAK/OSR1 kinases in controlling chloride influx
Much work in Dario Alessi's lab over the last 10 years has focused on understanding the function and regulation of the WNK1 and WNK4 family of protein kinases whose overexpression in humans causes Gordon's hypertension syndrome. Patients with this condition suffer from high blood pressure and hyperkalemia (high serum potassium), and can be treated using thiazide diuretic hypertension drugs that inhibit the NCC ion co-transporter in the kidney.
Work from the Alessi lab, as well as other labs, has revealed that the WNK enzymes are activated in response to hyperosmotic stress such as hypotonic low chloride conditions and phosphorylate and activate two other closely related protein kinases termed SPAK and OSR1. Once activated SPAK and OSR1 promote chloride influx into cells by phosphorylating and stimulate the activity of 3 cation-Cl- co-transporters collectively termed N[K]CCs which comprise NKCC1 (expressed in all cells), NCC or NKCC2 (expressed in the kidney).
Mutations that increase expression of WNK1 and WNK4 therefore cause hypertension by inducing too much activation of the SPAK/OSR1 kinases, which in turn result in over-stimulation of NCC and NKCC2 leading to inappropriately high salt retention in the kidnet. This also explains why thiazide diuretics that inhibit NCC are so effective at lowering blood pressure in Gordon syndrome patients.
It turns out that although N[K]CCs play a critical role in regulating intracellular chloride concentration –enzymes that catalyse the opposite reaction namely those controlling chloride efflux also play an important role. The enzymes that control chloride efflux are called KCCs and are also cation-Cl- co-transporters.
Much previous work has suggested that the activity of N[K]CCs and KCCs are coordinately regulated. ie conditions that stimulate net chloride influx (e.g. hypotonic low chloride) activate N[K]CCs and inhibit KCCs. Vice versa conditions that stimulate net chloride eflux (e.g. hypotonic high potassium) inhibit N[K]CCs and activate KCCs. Moreover, as discussed above phosphorylation activates N[K]CCs, but in contrast, phosphorylation by unknown kinases inhibits KCCs. Conversely, dephosphorylation inhibits N[K]CCs but activates KCCs. This reciprocal and opposite regulation of Na+- and K+-driven CCCs - ensures that cellular Cl- influx and efflux is tightly coordinated and this mechanism is conserved from worms to man.
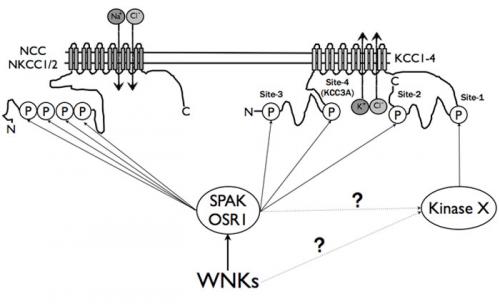
Previous seminal work by Jesse Rinehart and Richard Lifton at Yale revealed that activity of KCCs is critically controlled by phosphorylation of two highly conserved residues (termed Site-1 and Site-2) located within the intracellular C-terminal domain of these enzymes.
The identify of the kinases phosphorylating these vital sites on KCC isoforms were not known. Paola de los Heros and Jinwei Zhang, postdocs in the Alessi lab, in collaboration with Kris Kahle at Harvard University therefore set out to search for kinases that regulate KCCs. In the first part of this work they have now excitingly discovered that it is the WNK-regulated SPAK/OSR1 kinases that directly phosphorylate Site-2 on all KCC isoforms, thereby leading to the inhibition of the K+-Cl- co-transporters. Firstly, Paola discovered that SPAK and OSR1, in the presence of the MO25 regulatory subunit, robustly phosphorylated all KCC isoforms at Site-2 in vitro. Secondly, Jinwei found that STOCK1S-50699, a WNK pathway inhibitor, suppressed SPAK/OSR1 activation and both overexpressed and endogenous KCC3A Site-2 phosphorylation with similar efficiency. Thirdly, in embryonic stem cells lacking SPAK/OSR1 activity Paola and Jinwei discovered that endogenous phosphorylation of KCC isoforms at Site-2 is abolished and these cells display elevated basal activity of 86Rb+. Finally Jinwei established that there was a tight correlation between SPAK/OSR1 activity and the magnitude of KCC3A Site-2 phosphorylation. Paola also observed that KCCs are directly phosphorylated by SPAK/OSR1, at a novel Site-3 (Thr5 KCC1/KCC3 and Thr6 KCC2/KCC4), and a previously recognised KCC3-specific residue, Site-4 (Ser96).
These data demonstrate the WNK-regulated SPAK/OSR1 kinases directly phosphorylate the N[K]CCs and KCCs, promoting their stimulation and inhibition, respectively.
Given these reciprocal actions with anticipated net effects of increasing chloride influx, the new data suggests that targeting of WNK-SPAK/OSR1 with kinase inhibitors might comprise a new strategy to enhance cellular chloride extrusion. This has potential implications for the therapeutic modulation of epithelial and neuronal ion transport in human disease states.
In future work it will be exciting to identify the kinase that regulates KCCs at Site-1 and work out whether this is regulated by WNK isoforms.
To read a copy of Paola and Jinwei's paper click here
Work from the Alessi lab, as well as other labs, has revealed that the WNK enzymes are activated in response to hyperosmotic stress such as hypotonic low chloride conditions and phosphorylate and activate two other closely related protein kinases termed SPAK and OSR1. Once activated SPAK and OSR1 promote chloride influx into cells by phosphorylating and stimulate the activity of 3 cation-Cl- co-transporters collectively termed N[K]CCs which comprise NKCC1 (expressed in all cells), NCC or NKCC2 (expressed in the kidney).
Mutations that increase expression of WNK1 and WNK4 therefore cause hypertension by inducing too much activation of the SPAK/OSR1 kinases, which in turn result in over-stimulation of NCC and NKCC2 leading to inappropriately high salt retention in the kidnet. This also explains why thiazide diuretics that inhibit NCC are so effective at lowering blood pressure in Gordon syndrome patients.
It turns out that although N[K]CCs play a critical role in regulating intracellular chloride concentration –enzymes that catalyse the opposite reaction namely those controlling chloride efflux also play an important role. The enzymes that control chloride efflux are called KCCs and are also cation-Cl- co-transporters.
Much previous work has suggested that the activity of N[K]CCs and KCCs are coordinately regulated. ie conditions that stimulate net chloride influx (e.g. hypotonic low chloride) activate N[K]CCs and inhibit KCCs. Vice versa conditions that stimulate net chloride eflux (e.g. hypotonic high potassium) inhibit N[K]CCs and activate KCCs. Moreover, as discussed above phosphorylation activates N[K]CCs, but in contrast, phosphorylation by unknown kinases inhibits KCCs. Conversely, dephosphorylation inhibits N[K]CCs but activates KCCs. This reciprocal and opposite regulation of Na+- and K+-driven CCCs - ensures that cellular Cl- influx and efflux is tightly coordinated and this mechanism is conserved from worms to man.
Previous seminal work by Jesse Rinehart and Richard Lifton at Yale revealed that activity of KCCs is critically controlled by phosphorylation of two highly conserved residues (termed Site-1 and Site-2) located within the intracellular C-terminal domain of these enzymes.
The identify of the kinases phosphorylating these vital sites on KCC isoforms were not known. Paola de los Heros and Jinwei Zhang, postdocs in the Alessi lab, in collaboration with Kris Kahle at Harvard University therefore set out to search for kinases that regulate KCCs. In the first part of this work they have now excitingly discovered that it is the WNK-regulated SPAK/OSR1 kinases that directly phosphorylate Site-2 on all KCC isoforms, thereby leading to the inhibition of the K+-Cl- co-transporters. Firstly, Paola discovered that SPAK and OSR1, in the presence of the MO25 regulatory subunit, robustly phosphorylated all KCC isoforms at Site-2 in vitro. Secondly, Jinwei found that STOCK1S-50699, a WNK pathway inhibitor, suppressed SPAK/OSR1 activation and both overexpressed and endogenous KCC3A Site-2 phosphorylation with similar efficiency. Thirdly, in embryonic stem cells lacking SPAK/OSR1 activity Paola and Jinwei discovered that endogenous phosphorylation of KCC isoforms at Site-2 is abolished and these cells display elevated basal activity of 86Rb+. Finally Jinwei established that there was a tight correlation between SPAK/OSR1 activity and the magnitude of KCC3A Site-2 phosphorylation. Paola also observed that KCCs are directly phosphorylated by SPAK/OSR1, at a novel Site-3 (Thr5 KCC1/KCC3 and Thr6 KCC2/KCC4), and a previously recognised KCC3-specific residue, Site-4 (Ser96).
These data demonstrate the WNK-regulated SPAK/OSR1 kinases directly phosphorylate the N[K]CCs and KCCs, promoting their stimulation and inhibition, respectively.
Given these reciprocal actions with anticipated net effects of increasing chloride influx, the new data suggests that targeting of WNK-SPAK/OSR1 with kinase inhibitors might comprise a new strategy to enhance cellular chloride extrusion. This has potential implications for the therapeutic modulation of epithelial and neuronal ion transport in human disease states.
In future work it will be exciting to identify the kinase that regulates KCCs at Site-1 and work out whether this is regulated by WNK isoforms.
To read a copy of Paola and Jinwei's paper click here