Characterisation of the first selective inhibitor of the Parkinson's disease LRRK2 kinase
Autosomal dominant missense mutations within the gene encoding for the Leucine-Rich Repeat protein Kinase 2 (LRRK2) predispose humans to develop Parkinson's disease (PD). Little is understood about how LRRK2 is regulated, or how it functions, or how mutations cause Parkinson's disease. The most frequent LRRK2 mutation (G2019S) enhances kinase activity suggesting inhibitors may be useful for the treatment of Parkinson's disease. Whether inhibiting LRRK2 could be used as a strategy to treat Parkinson's disease is unknown and a key question for research in this field.
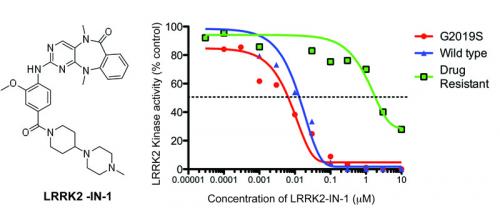
Nic Dzamko working in collaboration with Xianming Deng and Nathanael Gray at the Dana-Farber Cancer Institute have characterised a new compound termed LRRK2-IN-1 that potently inhibits LRRK2 (see figure). This compound was generated in the Gray laboratory and inhibits wild type LRRK2 with an IC50 of 13 nM and LRRK2[G2019S] mutant with an IC50 of 6 nM.
LRRK2 specificity was comprehensively evaluated against over 470 protein kinases and found to be remarkably specific, inhibiting only one other kinase namely MAPK7/ERK5 with similar affinity to LRRK2. Through molecular modelling of how LRRK2-IN-1 may bind to LRRK2, we were able to elaborate a LRRK2 mutation (A2016T) which is normally active, but ~400-fold resistant to LRRK2-IN-1.
As no LRRK2 substrates have yet been identified, there was no easy way to evaluate how effectively LRRK2-IN-1 suppressed LRRK2 activity in vivo. However, previous work undertaken by Nicolas Dzamko and a former postdoctoral fellow Jeremy Nichols (now a Principal Investigator, The Parkinson's Institute, Sunnyvale, California) suggested that phosphorylation of LRRK2 at two residues (Ser910 and Ser935) was controlled by a feedback pathway regulated by LRRK2 kinase activity itself [1, 2].
If this is correct, then LRRK2-IN-1 should induce dephosphorylation of endogenous LRRK2 at Ser910 and Ser935. Nic therefore tested the effect that LRRK2-IN-1 had on Ser910 and Ser935 phosphorylation in various cell types. He found, consistent with our hypothesis, that LRRK2-IN-1 promoted rapid dephosphorylation of Ser910 and Ser935 on endogenous LRRK2. Nic also found that injection of LRRK2-IN-1 into mice induced dephosphorylation of LRRK2 in the kidney and spleen, but not in the brain, indicating that LRRK2-IN-1 poorly penetrates the blood-brain barrier.
Previous work also suggested that Ser910 and Ser935 phosphorylation promoted binding of 14-3-3 and maintained LRRK2 diffusely localised in the cytoplasm of cells [1, 2]. Consistent with this, Nic found that treatment of cells with LRRK2-IN-1 induced dissociation of 14-3-3 binding and promoted the accumulation of LRRK2 within aggregate-like structures in the cytoplasm.
Importantly, LRRK2-IN-1 did not induce dephosphorylation or inhibit 14-3-3 binding or induce aggregation of the drug resistant LRRK2[A2016T] mutant. We would recommend that for all work undertaken with LRRK2-IN1, the drug resistant mutant be employed as a control to ensure effects of LRRK2-IN1 are not mediated inhibition of other targets such as ERK5.
These data confirm that LRRK2-IN1 does indeed effectively inhibit LRRK2 activity in vivo. These results provide further evidence to support the hypothesis that phosphorylation of Ser910 and Ser935 is controlled by LRRK2 kinase activity. We recommend that monitoring phosphorylation of LRRK2 at Ser910 and Ser935 be employed to assess the effectiveness of LRRK2 inhibitors being developed by the pharmaceutical industry. It will also be important to identify the protein kinase that phosphorylates Ser910 and Ser935 and determine whether it is regulated by LRRK2.
The discovery of LRRK2-IN-1 is important, as it will be a very useful tool compound to explore the physiological roles of LRRK2. We hope that LRRK2-IN1 could have a similar impact to the LRRK2 research field as other kinase inhibitors such as PD98059, SB203580 and wortmanin/LY294002 have had on elucidating the roles of the ERK, P38 and PI 3-kinase signalling pathways. We will make LRRK2-IN-1 available with no strings attached to any laboratory working on LRRK2. Hopefully LRRK2-IN-1 will also help with defining whether inhibiting LRRK2 would have utility for the treatment of Parkinson's disease.
It is also noteworthy that LRRK2-IN-1 was generated in the academic laboratory of Nathanael Gray, who has also developed hugely specific and potent inhibitors to many other important cellular kinases including mTOR, Abl, EGFR, FGFR, Eph receptor, ERK5 and aurora.
There is the perception that pharmaceutical companies can only generate highly specific kinase inhibitors, and that academics are best to stay out of this business. However, the work that Nathanael Gray has undertaken demonstrates that it is indeed possible to elaborate very useful tool compounds with the potential to revolutionise understanding of biological system outside a pharmaceutical environment.
It is likely that many of the future advances in our understanding of biological systems will be made possible by the elaboration of new chemical tools such as LRRK2-IN-1 and therefore medicinal chemistry whether undertaken in company or academic setting deserves great support.
To Read the recent LRRK2-IN-1 paper click here
References
1 Nichols, J., Dzamko, N., Morrice, N. A., Campbell, D. G., Deak, M., Ordureau, A., Macartney, T., Tong, Y., Shen, J., Prescott, A. and Alessi, D. R. (2010) 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson's disease associated mutations and regulates cytoplasmic localisation. Biochem J. 430, 393-404
2 Dzamko, N., Deak, M., Henati, F., Reith, A. D., Prescott, A. R., Alessi, D. R. and Nichols, R. J. (2010) Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser910/Ser935, disruption of 14-3-3 binding and altered cytoplasmic localisation. Biochem J 430, 405-413
Nic Dzamko working in collaboration with Xianming Deng and Nathanael Gray at the Dana-Farber Cancer Institute have characterised a new compound termed LRRK2-IN-1 that potently inhibits LRRK2 (see figure). This compound was generated in the Gray laboratory and inhibits wild type LRRK2 with an IC50 of 13 nM and LRRK2[G2019S] mutant with an IC50 of 6 nM.
LRRK2 specificity was comprehensively evaluated against over 470 protein kinases and found to be remarkably specific, inhibiting only one other kinase namely MAPK7/ERK5 with similar affinity to LRRK2. Through molecular modelling of how LRRK2-IN-1 may bind to LRRK2, we were able to elaborate a LRRK2 mutation (A2016T) which is normally active, but ~400-fold resistant to LRRK2-IN-1.
As no LRRK2 substrates have yet been identified, there was no easy way to evaluate how effectively LRRK2-IN-1 suppressed LRRK2 activity in vivo. However, previous work undertaken by Nicolas Dzamko and a former postdoctoral fellow Jeremy Nichols (now a Principal Investigator, The Parkinson's Institute, Sunnyvale, California) suggested that phosphorylation of LRRK2 at two residues (Ser910 and Ser935) was controlled by a feedback pathway regulated by LRRK2 kinase activity itself [1, 2].
If this is correct, then LRRK2-IN-1 should induce dephosphorylation of endogenous LRRK2 at Ser910 and Ser935. Nic therefore tested the effect that LRRK2-IN-1 had on Ser910 and Ser935 phosphorylation in various cell types. He found, consistent with our hypothesis, that LRRK2-IN-1 promoted rapid dephosphorylation of Ser910 and Ser935 on endogenous LRRK2. Nic also found that injection of LRRK2-IN-1 into mice induced dephosphorylation of LRRK2 in the kidney and spleen, but not in the brain, indicating that LRRK2-IN-1 poorly penetrates the blood-brain barrier.
Previous work also suggested that Ser910 and Ser935 phosphorylation promoted binding of 14-3-3 and maintained LRRK2 diffusely localised in the cytoplasm of cells [1, 2]. Consistent with this, Nic found that treatment of cells with LRRK2-IN-1 induced dissociation of 14-3-3 binding and promoted the accumulation of LRRK2 within aggregate-like structures in the cytoplasm.
Importantly, LRRK2-IN-1 did not induce dephosphorylation or inhibit 14-3-3 binding or induce aggregation of the drug resistant LRRK2[A2016T] mutant. We would recommend that for all work undertaken with LRRK2-IN1, the drug resistant mutant be employed as a control to ensure effects of LRRK2-IN1 are not mediated inhibition of other targets such as ERK5.
These data confirm that LRRK2-IN1 does indeed effectively inhibit LRRK2 activity in vivo. These results provide further evidence to support the hypothesis that phosphorylation of Ser910 and Ser935 is controlled by LRRK2 kinase activity. We recommend that monitoring phosphorylation of LRRK2 at Ser910 and Ser935 be employed to assess the effectiveness of LRRK2 inhibitors being developed by the pharmaceutical industry. It will also be important to identify the protein kinase that phosphorylates Ser910 and Ser935 and determine whether it is regulated by LRRK2.
The discovery of LRRK2-IN-1 is important, as it will be a very useful tool compound to explore the physiological roles of LRRK2. We hope that LRRK2-IN1 could have a similar impact to the LRRK2 research field as other kinase inhibitors such as PD98059, SB203580 and wortmanin/LY294002 have had on elucidating the roles of the ERK, P38 and PI 3-kinase signalling pathways. We will make LRRK2-IN-1 available with no strings attached to any laboratory working on LRRK2. Hopefully LRRK2-IN-1 will also help with defining whether inhibiting LRRK2 would have utility for the treatment of Parkinson's disease.
It is also noteworthy that LRRK2-IN-1 was generated in the academic laboratory of Nathanael Gray, who has also developed hugely specific and potent inhibitors to many other important cellular kinases including mTOR, Abl, EGFR, FGFR, Eph receptor, ERK5 and aurora.
There is the perception that pharmaceutical companies can only generate highly specific kinase inhibitors, and that academics are best to stay out of this business. However, the work that Nathanael Gray has undertaken demonstrates that it is indeed possible to elaborate very useful tool compounds with the potential to revolutionise understanding of biological system outside a pharmaceutical environment.
It is likely that many of the future advances in our understanding of biological systems will be made possible by the elaboration of new chemical tools such as LRRK2-IN-1 and therefore medicinal chemistry whether undertaken in company or academic setting deserves great support.
To Read the recent LRRK2-IN-1 paper click here
References
1 Nichols, J., Dzamko, N., Morrice, N. A., Campbell, D. G., Deak, M., Ordureau, A., Macartney, T., Tong, Y., Shen, J., Prescott, A. and Alessi, D. R. (2010) 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson's disease associated mutations and regulates cytoplasmic localisation. Biochem J. 430, 393-404
2 Dzamko, N., Deak, M., Henati, F., Reith, A. D., Prescott, A. R., Alessi, D. R. and Nichols, R. J. (2010) Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser910/Ser935, disruption of 14-3-3 binding and altered cytoplasmic localisation. Biochem J 430, 405-413