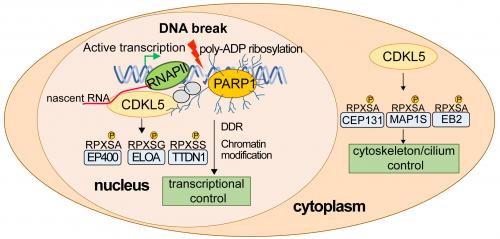
Mutations in the gene encoding the CDKL5 kinase are among the most common genetic causes of childhood epilepsy and can also give rise to the severe neurodevelopmental condition CDD (CDKL5 Deficiency Disorder). Despite its importance for human health, the phospho–targets and cellular roles of CDKL5 are poorly understood. Finding the cellular substrates of CDKL5 is critically important because the pathological mutations in CDKL5 reduce kinase activity to varying degrees, implying that is a reduction in substrate phosphorylation that causes brain dysfunction and neurological disease. The Rouse team and others reported the first physiological substrates of CDKL5 in 2018, which are involved in control of the microtubule-based cytoskeleton in the cytoplasm. However, CDKL5 is also located in the nucleus but little is known about its functions in this compartment, and nuclear phospho–targets were conspicuously absent from the screens reported in 2018.
Today in the EMBO Journal, a study funded by the Medical Research Council (MRC) led by researchers Taran Khanam and Ivan Munõz in John Rouse’s team at the MRC PPU, with help from Florian Weiland (now at KU Leuven), Houjiang Zhou (now at Zhejiang Hisun Pharmaceutical Co., LTD) and other MRC PPU researchers and support staff, reports a key role for CDKL5 in the nucleus. The report describes the identification of CDKL5 in a cell-based screen for kinases recruited to sites of DNA damage; intriguingly, CDKL5 is recruited to DNA breaks that occur in regions of ongoing transcription. The recruitment of CDKL5 to DNA damage sites requires the local synthesis of a nucleic acid-like polymer called poly (ADP-ribose), catalysed by poly (ADP-ribose) polymerase 1 (PARP1), and also requires nascent mRNA synthesis. It therefore appears that CDKL5 acts as a coincidence detector recognizing breaks (and maybe other lesions) that occur at sites of active transcription.
In the same report, the authors reveal how quantitative phospho-proteomic screening identified the first physiological nuclear substrates of CDKL5, revealing a network of transcriptional regulators including Elongin A (ELOA), phosphorylated on a specific CDKL5 consensus motif. Recruitment of CDKL5 and ELOA to damaged DNA, and subsequent phosphorylation of ELOA, requires both active transcription and the synthesis of poly–ADP ribose, to which CDKL5 can bind. These data suggest a model where a common recruitment mechanism involving poly ADP ribosylation and nascent RNA synthesis juxtaposes kinase and substrate at DNA breaks, enabling robust phosphorylation of substrate by kinase.
The requirement for transcription in CDKL5 recruitment to DNA breaks, and the enrichment of transcriptional regulators in the substrate screen, pointed strongly to a role in modulating transcription at DNA breaks. A well–documented response to DNA lesions such as double-strand breaks is the silencing of transcription near the lesion, presumably to facilitate access to repair proteins and proteins that reset chromatin status before and after repair. In the new study CDKL5 kinase activity was found to be required for the silencing of genes harboring DNA breaks, in work carried out in collaboration with researchers in the lab of Prof. Tibor Pankotai at the University of Szeged, Hungary. This finding has implications for understanding the molecular basis of CDKL5-related diseases, as defects in the nuclear functions of CDKL5 may be crucial for understanding the molecular basis of CDD. Moreover, the Identification of CDKL5 substrates also paves the way for the development of rational therapies to treat CDKL5-based diseases. Cell-based assays employing phospho–specific antibodies against CDKL5 targets could be used screen for small molecules or gene deletions that rescue phosphorylation of these targets in human disease.